Medical Management & Healthcare · 7 months ago
The Best Medical Management of Androgen Insensitivity Syndrome (AIS)
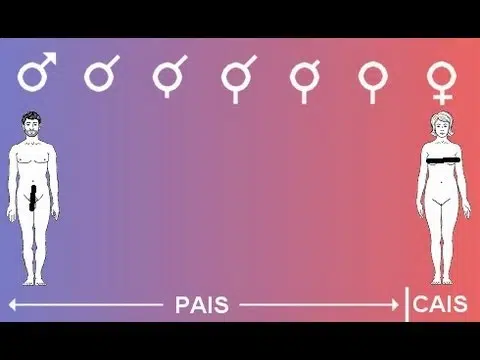
Androgen Insensitivity Syndrome (AIS) is a complex genetic condition that requires thoughtful, individualized, and ethically grounded medical care. In 2026,…